Recent News
We are looking for healthy blood donors for immunological research
Currently, The Department of Genome Informatics, Research Institute for Microbial Diseases, Osaka University is looking for healthy people who can provide blood samples for immunology research. If you would like to cooperate with blood donation (blood collection), please contact us. After hearing about your health condition, we will collect a small amount (less than 50 ml) of blood. We will compare the properties of immune cells of healthy and sick people, and use them for AI development useful for medical treatment and research.
Target people:
Healthy adults who have been vaccinated with DTP (Diphtheria, Tetanus, Pertussis) Booster Vaccination in the past or willing to be vaccinated in the near future. We will listen to your health condition, explain your participation in the research, ask for consent (informed consent), and collect blood at the Immunology Frontier Research Center, Osaka University. Blood is collected by the usual method, and less than 50 ml of blood is collected.
Privacy protection:
The data used for research and publication in academic papers will be anonymized and managed in a format so that it cannot be identified by an individual.
Research subject name:
Human immune repertoire analysis using single cell analysis and development of epitope prediction using AI system
Purpose of research:
Sequence analysis is performed to simultaneously clarify the immune receptor sequence, gene expression, and antigen epitope information of T cells or B cells, and identify antigens and receptors that are important in medical applications. We will develop an AI system by using the immunoreceptor sequence information obtained by the experiment and the correspondence information of the antigen epitope (not including personal information) as learning data together with other public data.
Principal investigator:
Daron Standley (Department of Genome Informatics, Osaka University)
Further inquiries:
Dianita Susilo (Department of Genome Informatics, Osaka University) Telephone: 06-6879-8368 E-mail: dianitasusilo@biken.osaka-u.ac.jp
Update on emerging SARS-CoV-2 variants
We previously investigated the evolutionary Importance of residue positions in the SARS-CoV-2 S protein (Saputri et al. Frontiers in Microbiology 2020). Here, the Importance of a site was defined as the difference in diversity in two groups of viruses
Importance = diversity(SARS-CoV-2+outgroup)−diversity(SARS-CoV-2),
Where outgroup included betacoronaviruses infecting bat and pangolin. We subsequently found that the Importance was a strong indicator of mutations in emerging variants with increased infectivity from England, South Africa and Brazil. We then noticed that diversity(outgroup) was the major contribution to Importance. Because of the potential use in predicting future variants, we here provide a complete list S protein diversity values. We list both the original definition of outgroup, as well as a slightly broader definition, in order to increase the amount of data. Regardless of which definition we chose, the correlation between diversity(outgroup) and emerging mutation is significant (Fisher's Exact Test).
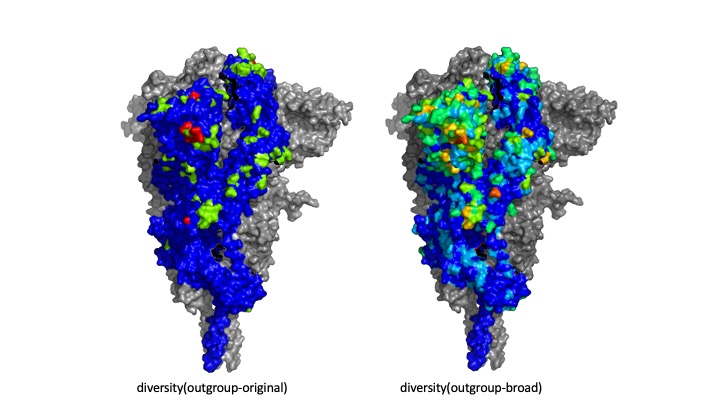
|
diversity(outgroup-original)
|
1
|
2
|
3
|
(p-value)
|
|---|---|---|---|---|
|
No. sites
|
1180
|
85
|
9
|
|
|
Mutations in variants
|
15
|
4
|
2
|
(0.00099)
|
|
Mutations in multiple variants
|
0
|
3
|
2
|
(1.4e-07)
|
|
diversity(outgroup-broad)
|
1
|
2
|
3
|
4 | 5 | 6 | 7 | (p-value) |
|---|---|---|---|---|---|---|---|---|
|
No. sites
|
822 | 242 | 126 | 57 | 23 | 2 | 1 | |
|
Mutations in variants
|
8 | 3 | 6 | 2 | 2 | 0 | 0 | (0.0057) |
|
Mutations in multiple variants
|
0 | 1 | 1 | 1 | 2 | 0 | 0 | (0.00046) |
Announcing Support for 2019-nCoV Sequence Analysis (日本語) (中国語)
The outbreak of a novel coronavirus (2019-nCoV) has prompted many scientists to share their findings through unconventional means such as social media, blogs and impromptu email threads. Recently, a post on Twitter identified more than 300 2019-nCoV-related papers on bioRxiv and requested support from the scientific community in vetting these preprints. Among the questions asked was a direct appeal to our lab to determine whether the MAFFT sequence alignment software was being used appropriately. In addition to attempting to respond to this specific question, we would like to add several general points as follows:
1. MAFFT is frequently used in MSA analyses of 2019-nCoV. For example, in the published literature:
- Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding; Lu, et al.; Lancet 2020
- Potential of large ‘first generation’ human-to-human transmission of 2019-nCoV; Li, et al.; Journal of Medical Virology 2020
- The 2019‐new coronavirus epidemic: evidence for virus evolution; Benvenuto, et al.; Journal of Medical Virology 2020
- Homologous recombination within the spike glycoprotein of the newly identified coronavirus...; Ji, et al.; Journal of Medical Virology 2020
It’s not easy for us to reproduce every MSA by collecting the same input sequences. Therefore, in order to both validate MSA methods and make these analyses more smooth, we propose to accept sequence data from anyone working on 2019-nCoV and, as long as we have time and our computational resources allow, appropriately apply MAFFT to prepare MSAs and/or trees.
2. In addition to carrying out analysis of virus sequences, it may be possible to obtain valuable information pertaining to the immune responses to 2019-nCoV by sequencing the B and T cell receptors of infected individuals. We can also accept such BCR or TCR repertoire data, and use our own in-house tools or third-party tools to identify likely 2019-nCoV-responsive BCRs or TCRs.
If you wish to have your sequence data analyzed in this fashion, please contact us using one of the forms below:
Do not include the data itself (or a link to the data) in this request. After receiving your inquiry we will ask for access to the data and further details.
Highly-Cited Papers in Web of Science (Top 0.1%)
- K Katoh, J Rozewicki, and KD Yamada "MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization", Briefings in Bioinformatics, vol. 20 no. 14 (2019)
Kazutaka Katoh & MAFFT in Nikkei Sangyo Shimbun
- Kazutaka Katoh, author of the MAFFT multiple alignment software, was recently profiled by a journalist from Nikkei Sangyo Shimbun.
Highly-Cited Papers in Web of Science (Top 1%)
- J Rozewicki, S Li, KM Amada, DM Standley and K Katoh, "MAFFT-DASH: integrated protein sequence and structural alignment", Nucleic Acids Research, no.47 (2019)
- T Nakamura, KD Yamada, K Tomii and K Katoh, "Parallelization of MAFFT for large-scale multiple sequence alignments", Bioinformatics 34, no.14 (2018)
- K Katoh and DM Standley, "A simple method to control over-alignment in the MAFFT multiple sequence alignment program", Bioinformatics 32, no.13 (2016)
