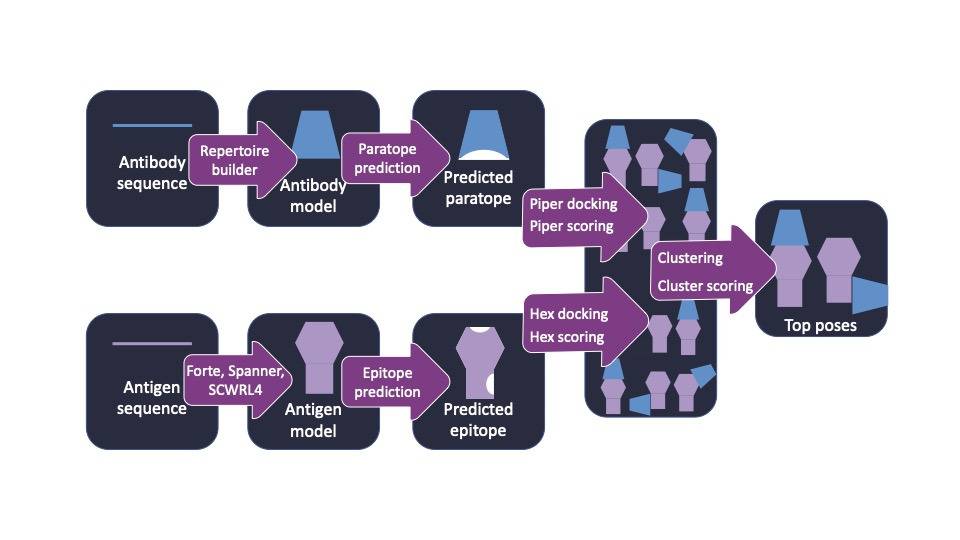
AbAdapt is an antibody-antigen docking pipeline that allows input of either sequences or structures. It integrates methods for structural modeling, binding site prediction, docking and scoring. The specific steps involved are described in detail here using an example calculation.
AbAdapt uses in-house tools for antibody and antigen modeling.
Antibody inputs. AbAdapt uses Repertoire Builder to render 3D structures of antibodies from sequence. Sequences should be entered in FASTA format, with heavy chains first and light chains second. The example on the AbAdapt web server is taken from the influenza neutralizing antibody L4B-18, which has a corresponding entry in the Protein data Bank (entry 6II8), which was deposited after the Repertoire Builder software was built. It thus provides an example for which the structure is known but the input models will not be unrealistically good. The example antibody sequence looks like this:
>6II8_G EVQLVESGGGLVQPGGSLRLSCAASGFTFSSYAMSWVRQAPGKGLEWVSAISGSGGSTYYADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCAKDRLLWFGELLFEGFDYWGQGTLVTVSS >6II8_I NFMLTQPHSVSESPGKTVTISCTRSSGSIASNYVQWYQQRPGSSPTTVIYEDNQRPSGVPDRFSGSIDSSSNSASLTISGLKTEDEADYYCQSYDSSNVVFGGGTKLTVL
Repertoire Builder exists as a stand-alone web server and has been described previously here. The key thing to be aware of is that Repertoire Builder uses template-based modeling. If there are no suitable templates for the CDRs in your antibody of interest, Repertoire Builder will either return a low accuracy model or possibly no model at all. For this reason, AbAdapt also allows structures to be input. Note that we have had success in using recently developed generic Deep Learning-based protein structure prediction software, such as AlphaFold for antibody modeling. If you do input an antibody structure, please take care to either label the heavy and light chains ‘H’ and ‘L’, respectively, or indicate the chain IDs (in the order heavy-light) on the web form. Adapt will re-label heavy and light chains ‘H’ and ‘L’ internally for all calculations. If needed, the chain IDs will be re-labeled back to the original values upon completion of the job.
Antigen Inputs. Antigen modeling is handled internally using the Spanner web server. Antigens that consist of multiple chains can be used. Using PDB ID 6II8 as an example, chains A and B would be input as follows:
>6II8_A DKICLGHHAVSNGTKVNTLTERGVEVVNATETVERTNIPRICSKGKRTVDLGQCGLLGTITGPPQCDQFLEFSADLIIERREGSDVCYPGKFVNEEALRQILRESGGIDKEAMGFTYSGIRTNGATSACRRSGSSFYAEMKWLLSNTDNAAFPQMTKSYKNTRKSPALIVWGIHHSVSTAEQTKLYGSGNKLVTVGSSNYQQSFVPSPGARPQVNGLSGRIDFHWLMLNPNDTVTFSFNGAFIAPDRASFLRGKSMGIQSGVQVDANCEGDCYHSGGTIISNLPFQNIDSRAVGKCPRYVKQRSLLLATGMKNVPE >6ii8_B GLFGAIAGFIENGWEGLIDGWYGFRHQNAQGEGTAADYKSTQSAIDQITGKLNRLIEKTNQQFELIDNEFNEVEKQIGNVINWTRDSITEVWSYNAELLVAMENQHTIDLADSEMDKLYERVKRQLRENAEEDGTGCFEIFHKCDDDCMASIRNNTYDHSKYREEAMQNRIQ
Regardless of whether sequence or structures are input, AbAdapt will label all antigen chains “A” internally. In such cases, the chain IDs will be re-labeled back to the original values upon completion of the job.
Reference PDB. If a reference complex is input, the errors in the docked poses will be computed relative to this reference. Actually, the reference is used indirectly: the antibody and antigen models are superimposed on the reference and this ‘idealized’ pose is used for all RMSD calculations. The reason is that this procedure guarantees that the residues and atoms (reference and model) will match in the RMSD calculations. However, the procedure can result in some distortions in the RMSD calculation, if local conformations of antibody, antigen or both exist. Note that the same considerations about chain IDs, described above, apply here: if the reference antibody (heavy-light) and antigen chains are not ‘HL’ and ‘A’, they will be re-labeled as such internally. AbAdapt will assume that any non-antibody chain in the reference is part of the antigen if the "antigen chain" input text box is blank.
Restraints. A restraint file is a list of antigen residues. It must match residues in the antigen sequence or input structure. The format is a space-delimited text file containing the following columns:
Using PDB entry 6II8 as an example, if we wanted to restrain residue Lysine 184 to be part of the epitope, we could upload a restraint file with the following line:
184 A K 1
You can add as many restraints to the file as you like (one per line). Note that the residue number and amino acid type must match to the input antigen. If a sequence is input, then the numbering and amino acid type should agree with the sequence, (starting from 1). If an antigen PDB file is uploaded, the numbering, chain ID and residue in the restraint file name should match that of the input PDB. Although AbAdapt internally re-labels antigen chains to “A”, the restraint file should match the original (i.e. the uploaded) input antigen. All the chain re-labeling is done internally.
Piper sampling frequency. This option controls the fraction of the 10,000 selected poses that are taken from Piper. A value of 1.0 means only Piper poses are used. The default value (0.9) means 9,000 are taken from Piper and 1,000 are taken from Hex.
Most obvious errors in the input should appear quickly as a message in a red textbox:

Since AbAdapt is a new tool, there are probably errors that will occur that we have not anticipated and will not trigger such clear error messages. If your results don’t appear within several hours, or anything strange happens, please report the problem to us using the feedback link at the bottom of the AbAdapt landing page, and we will try to fix the problem quickly.
Sample output can be loaded from the AbAdapt landing page. There are three main sections to this page: Result Files, Viewer, Clusters.
Result Files. The Result files include:
Viewer. We use the molmil molecular viewer which renders the antibody and antigen cluster representative colored by paratope and epitope probability.
Clusters. All clusters are shown in the table, and can be viewed in the viewer by clicking on the “View” link. The individual cluster representatives can also be downloaded. The cluster sizes and scores are indicated in the table. Here, the score is the final score that is based on Piper-Hex co-clustering.
© 2021 Department of Genome Informatics; Research Institute for Microbial Diseases; Osaka University